

Drug-drug Interaction Prediction Method Based on Multi-level Attention Mechanism and Message Passing Neural Network
-
摘要: 药物相互作用(Drug-drug interaction, DDI)是指不同药物存在抑制或促进等作用. 现有DDI预测方法往往直接利用药物分子特征表示预测DDI, 而忽略药物分子中不同原子对DDI的影响. 为此, 提出基于多层次注意力机制和消息传递神经网络的DDI预测方法. 该方法将DDI建模为通过学习基于序列表示的药物分子特征实现DDI预测的链接预测问题. 首先, 建立基于注意力机制和消息传递神经网络的原子特征网络, 结合提出的基于分子质心的位置编码, 学习不同原子及其相关联化学键的特征, 构建基于图结构的药物分子特征表示; 然后, 设计基于注意力机制的分子特征网络, 并通过监督和对比损失学习, 实现DDI预测; 最后, 通过实验证明该方法的有效性和优越性.Abstract: Drug-drug interaction (DDI) denotes the presence of inhibitory or promoting effects between different drugs. The existing DDI prediction methods often directly use drug molecular feature representation, while ignoring the different effects of different atoms within drug molecule on DDI. To solve this problem, a DDI prediction method is proposed based on multi-level attention mechanism and message passing neural network. This method models the task as a link prediction problem of predicting DDI by extracting the drug molecular features from their sequence representations. First, the atomic feature network is developed based on attention mechanism and message passing neural network. Through integration with the proposed positional encoding based on molecular centroid, the proposed network can learn from different atoms and the correlated chemical bonds to construct drug molecular graph features. Second, attention mechanism-based molecular feature network is designed, and the DDI prediction can then be realized by using supervision and contrastive loss learning. Finally, experiments demonstrate the effectiveness and superiority of the proposed method.
-
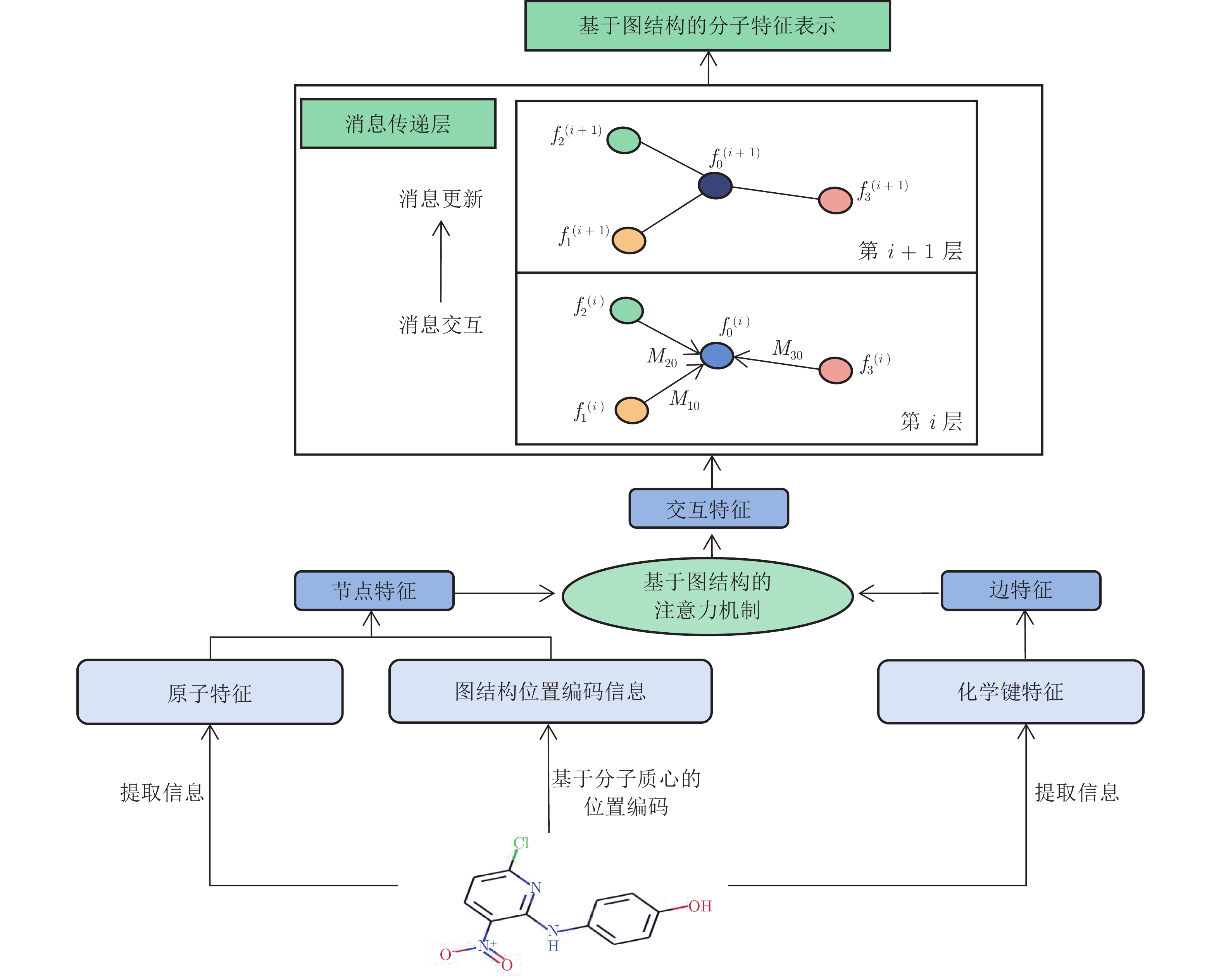
图 2 基于注意力机制的消息传递原子特征网络
Fig. 2 Framework of the message passing atomic feature network base on attention mechanism
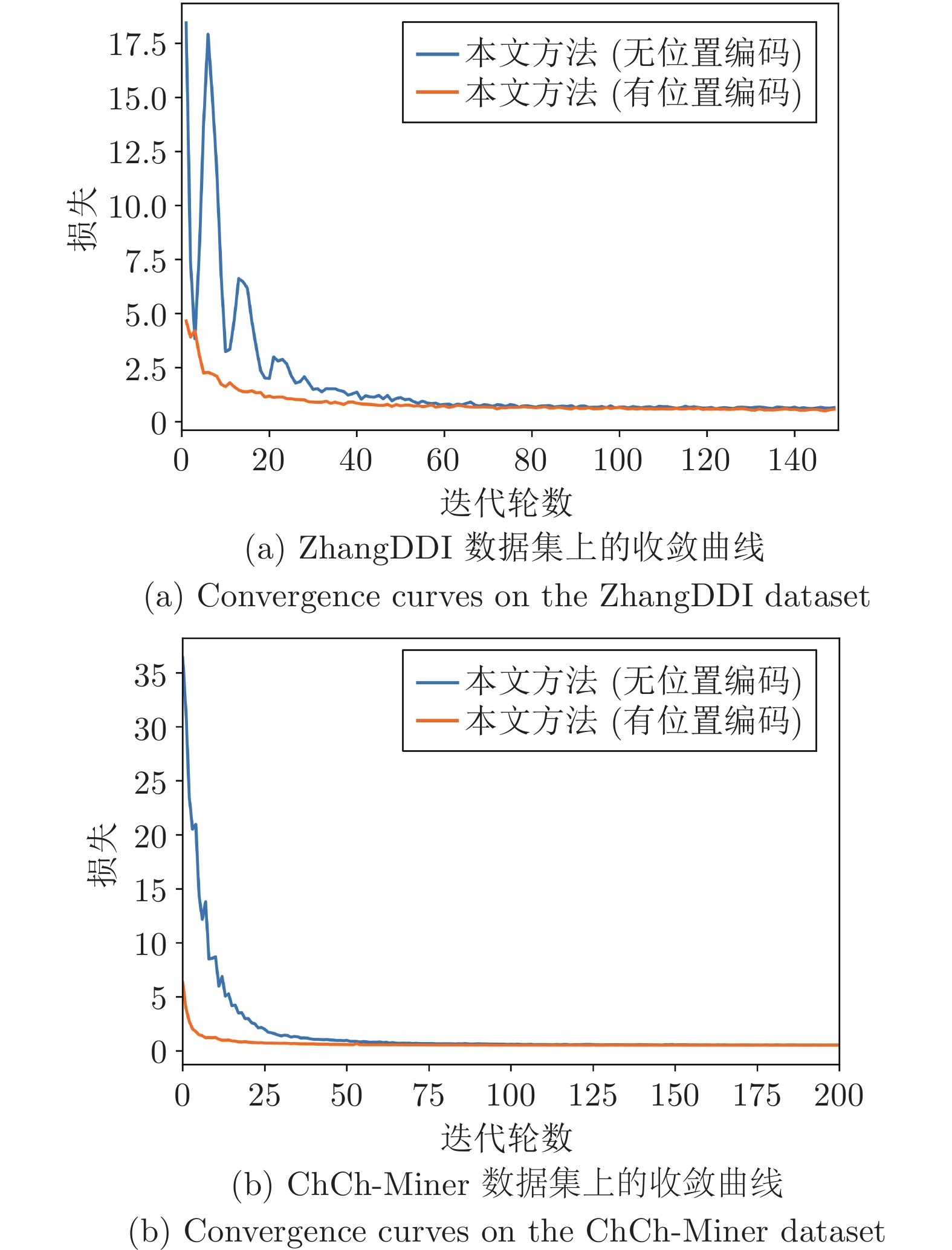
图 4 位置编码对模型收敛性能的影响
Fig. 4 The effect of positional encoding on model convergence performance
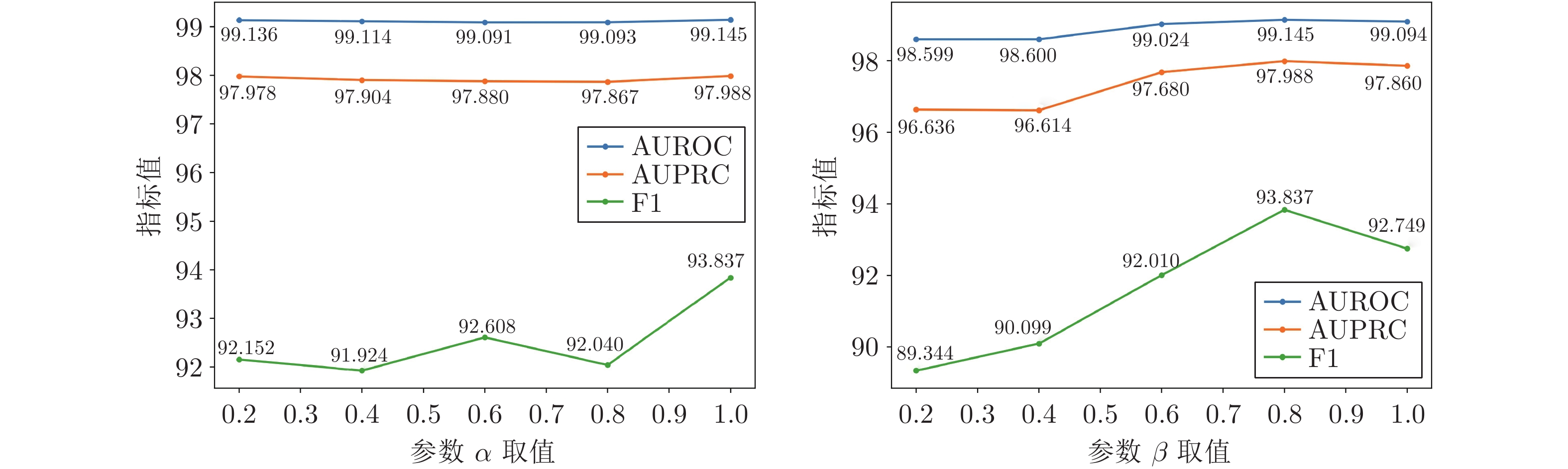
图 5 在ZhangDDI数据集上不同$\alpha$和$\beta$取值对模型性能的影响
Fig. 5 The effects of different $\alpha$ and $\beta$ on model performance on ZhangDDI dataset
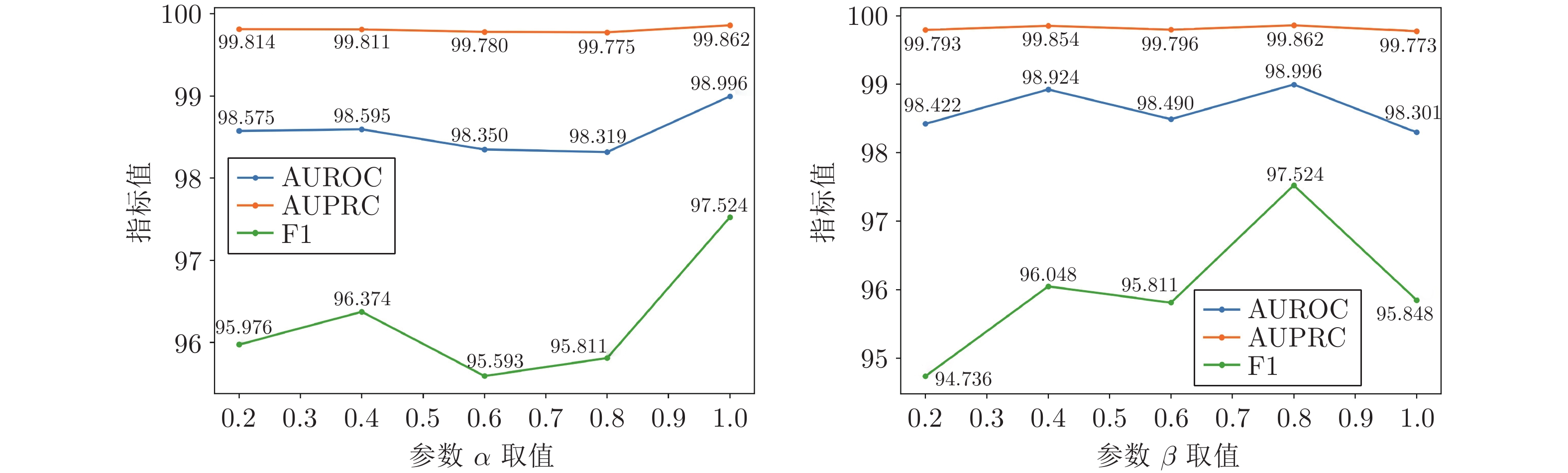
图 6 在ChCh-Miner数据集上不同$\alpha$和$\beta$取值对模型性能的影响
Fig. 6 The effects of different $\alpha$ and $\beta$ on model performance on ChCh-Miner dataset
表 1 ZhangDDI数据集上的对比实验结果
Table 1 Comparison experimental results on ZhangDDI dataset
模型 AUROC AUPRC F1 NN[24] 67.81±0.25 52.61±0.27 49.84±0.43 LP-Sub[25] 93.39±0.13 89.15±0.13 79.61±0.16 LP-SE[25] 93.48±0.25 89.61±0.19 79.83±0.61 LP-OSE[25] 93.50±0.24 90.31±0.82 80.41±0.51 MF-Ens[11] 95.20±0.14 92.51±0.15 85.41±0.16 SSP-MLP[1] 92.51±0.15 88.51±0.66 80.69±0.81 GCN[26] 91.91±0.62 88.73±0.84 81.61±0.39 GIN[27] 81.45±0.26 77.16±0.16 64.15±0.16 Att-auto[12] 92.84±0.61 90.21±0.19 70.96±0.39 GAT[28] 91.49±0.29 90.69±0.10 80.93±0.25 SEAL-CI[29] 92.93±0.19 92.82±0.17 84.74±0.17 NFP-GCN[30] 93.22±0.09 93.07±0.46 85.29±0.38 MIRACLE[13] 98.95±0.15 98.17±0.06 93.20±0.27 本文方法 99.14±0.01 97.97±0.02 93.79±0.28  下载: 导出CSV
下载: 导出CSV
表 3 原子特征网络的消融实验结果
Table 3 Ablation experimental results on atomic feature network
数据集 算法 AUROC AUPRC F1 ZhangDDI 无注意力的
原子网络98.70±0.20 96.89±0.50 90.46±1.18 本文方法 99.14±0.01 97.97±0.02 93.79±0.28 ChCh-Miner 无注意力的
原子网络95.90±0.99 99.18±0.15 96.23±0.34 本文方法 98.45±0.31 99.79±0.04 96.51±0.84
下载: 导出CSV
表 4 分子特征网络的消融实验结果
Table 4 Ablation experimental results on molecular feature network
数据集 算法 AUROC AUPRC F1 ZhangDDI 无注意力的
分子网络98.82±0.27 97.18±0.68 91.60±1.84 本文方法 99.14±0.01 97.97±0.02 93.79±0.28 ChCh-Miner 无注意力的
分子网络95.78±1.29 99.19±0.38 95.19±1.45 本文方法 98.45±0.31 99.79±0.04 96.51±0.84
下载: 导出CSV
表 5 位置编码对模型性能影响的对比结果
Table 5 Comparison results of the impact of positional encoding on model performance
数据集 算法 AUROC AUPRC F1 ZhangDDI 无位置编码 98.91±0.26 97.46±0.60 91.38±2.11 传统位置编码 99.02±0.21 97.68±0.53 92.70±1.35 本文方法 99.14±0.01 97.97±0.02 93.79±0.28 ChCh-Miner 无位置编码 95.62±2.79 99.11±0.63 96.12±0.58 传统位置编码 97.54±0.46 99.66±0.06 94.73±0.54 本文方法 98.45±0.31 99.79±0.04 96.51±0.84
下载: 导出CSV
表 6 损失函数对模型性能影响的对比结果
Table 6 Comparison results of the impact of loss function on model performance
数据集 算法 AUROC AUPRC F1 ZhangDDI 无自蒸馏约束项 98.71±0.01 96.87±0.02 89.53±0.54 无对比学习损失项 94.19±0.06 79.62±0.31 73.59±0.39 infoNCE 对比损失项 99.10±0.05 97.91±0.09 92.87±0.60 不同采样方式的对比损失项 99.13±0.02 97.97±0.04 93.15±0.61 本文方法 99.14±0.01 97.97±0.02 93.79±0.28 ChCh-Miner 无自蒸馏约束项 97.55±3.24 99.48±0.87 96.34±1.01 无对比学习损失项 58.70±5.00 90.30±1.06 94.89±0.57 infoNCE 对比损失项 98.59±0.20 99.80±0.03 97.31±0.20 不同采样方式的对比损失项 98.38±0.01 99.78±0.00 95.67±0.09 本文方法 98.45±0.31 99.79±0.04 96.51±0.84
下载: 导出CSV
-
[1] Ryu J Y, Kim H U, Lee S Y. Deep learning improves prediction of drug-drug and drug-food interactions. The National Academy of Sciences (NAS), 2018, 115(18): E4304−E4311 [2] Sun M Y, Zhao S D, Gilvary C, Elemento O, Zhou J Y, Wang F. Graph convolutional networks for computational drug development and discovery. Briefings in Bioinformatics, 2020, 21(3): 919-935 doi: 10.1093/bib/bbz042 [3] Qiu Y, Zhang Y, Deng Y F, Liu S C, Zhang W. A comprehensive review of computational methods for drug-drug interaction detection. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2022, 19(4): 1968−1985 [4] Abbas K, Abbasi A, Dong Shi, Niu L, Yu L H, Chen B, et al. Application of network link prediction in drug discovery. BMC Bioinformatics, 2021, 22(1): 1-21 doi: 10.1186/s12859-020-03881-z [5] 侯美好. 基于图神经网络的药物不良相互作用预测 [硕士学位论文], 山东大学, 中国, 2020.Hou Mei-Hao. Prediction of Adverse Drug Interactions Based on Graph Neural Network [Master thesis], Shandong University, China, 2020. [6] Lee G, Park C, Ahn J. Novel deep learning model for more accurate prediction of drug-drug interaction effects. BMC Bioinformatics, 2019, 20(1): 1-8 doi: 10.1186/s12859-018-2565-8 [7] Karim M R, Cochez M, Jares J B, Uddin M, Beyan O, Decker S. Drug-drug interaction prediction based on knowledge graph embeddings and convolutional-lstm network. In: Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics. New York, USA: ACM, 2019. 113−123 [8] Yan C, Duan G H, Zhang Y Y, Wu F X, Pan Y, Wang J X. IDNDDI: An integrated drug similarity network method for predicting drug-drug interactions. In: Proceedings of the 15th International Symposium on Bioinformatics Research and Applications. Cham, Switzerland: Springer, 2019. 89−99 [9] Liu S C, Huang Z Y, Qiu Y, Chen Y P P, Zhang W. Structural network embedding using multi-modal deep auto-encoders for predicting drug-drug interactions. In: Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine. San Diego, USA: IEEE, 2019. 445−450 [10] Deng Y F, Xu X R, Qiu Y, Xia J B, Zhang W, Liu S C. A multimodal deep learning framework for predicting drug–drug interaction events.Bioinformatics, 2020, 36(15): 4316-4322 doi: 10.1093/bioinformatics/btaa501 [11] Zhang W, Chen Y L, Liu F, Luo F, Tian G, Li X H. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data.BMC Bioinformatics, 2017, 18(1): 1-12 doi: 10.1186/s12859-016-1414-x [12] Ma T F, Xiao C, Zhou J Y, Wang F. Drug similarity integration through attentive multi-view graph auto-encoders. In: Proceedings of the 27th International Joint Conference on Artificial Intelligence. Stockholm, Sweden: Morgan Kaufmann, 2018. 3477−3483 [13] Wang Y H, Min Y S, Chen X, Wu J. Multi-view graph contrastive representation learning for drug-drug interaction prediction. In: Proceedings of the Web Conference (WWW'21). New York, USA: ACM, 2021. 2921−2933 [14] Yan C, Duan G H, Zhang Y Y, Wu F X, Pan Y, Wang J X. Predicting drug-drug interactions based on integrated similarity and semi-supervised learning.IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2022, 19(1): 168-179 doi: 10.1109/TCBB.2020.2988018 [15] Jin B, Yang H Y, Xiao C, Zhang P, Wei X P, Wang F. Multitask dyadic prediction and its application in prediction of adverse drug-drug interaction. In: Proceedings of the AAAI Conference on Artificial Intelligence. Palo Alto, USA: AAAI, 2017. 367−373 [16] Lin X, Quan Z, Wang Z J, Ma T F, Zeng X X. KGNN: Knowledge graph neural network for drug-drug interaction prediction. In: Proceedings of the 29th International Joint Conference on Artificial Intelligence. Yokohama, Japan: Morgan Kaufmann, 2020. 2739−2745 [17] Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, et al. Attention is all you need. In: Proceedings of the 30th Annual Conference on Neural Information Processing Systems. Barcelona, Spain: Curran Associates, 2017. 5998−6008 [18] Chen J W, Zheng S J, Song Y, Rao J H, Yang Y D. Learning attributed graph representations with communicative message passing transformer. In: Proceedings of the 30th International Joint Conference on Artificial Intelligence. Montreal, Canada: Morgan Kaufmann, 2021. 2242−2248 [19] Landrum G. Rdkit documentation [Online], available: https://www.rdkit.org/, July 29, 2022 [20] Gilmer J, Schoenholz S S, Riley P F, Vinyals O, Dahl G E. Neural message passing for quantum chemistry. In: Proceedings of the 34th International Conference on Machine Learning. Sydney, Australia: ACM, 2017. 1263−1272 [21] Zhang L F, Song J B, Gao A, Chen J W, Bao C L, Ma K S. Be your own teacher: Improve the performance of convolutional neural networks via self-distillation. In: Proceedings of the IEEE/CVF International Conference on Computer Vision. Seoul, South Korea: IEEE, 2019. 3713−3722 [22] Hjelm R D, Fedorov A, Lavoie-Marchildon S, Grewal K, Bachman P, Trischler A, et al. Learning deep representations by mutual information estimation and maximization. In: Proceedings of the 7th International Conference on Learning Representations. New Orleans, USA: OpenReview.net, 2019. [23] Zitnik M, Sosic R, Leskovec J. BioSNAP Datasets: Stanford biomedical network dataset collection [Online], available: http://snap.stanford.edu/biodata, July 30, 2022 [24] Vilar S, Harpaz R, Uriarte E, Santana L, Rabadan R, Friedman C. Drug—drug interaction through molecular structure similarity analysis. Journal of the American Medical Informatics Association, 2012, 19(6): 1066-1074 doi: 10.1136/amiajnl-2012-000935 [25] Zhang P, Wang F, Hu J Y, Sorrentino R. Label propagation prediction of drug-drug interactions based on clinical side effects. Scientific Reports, 2015, 5(1): 1-10 doi: 10.9734/JSRR/2015/14076 [26] Kipf T N, Welling M. Semi-supervised classification with graph convolutional networks. In: Proceedings of the 5th International Conference on Learning Representations. Toulon, France: OpenReview.net, 2017. [27] Xu K Y L, Hu W H, Leskovec J, Jegelka S. How powerful are graph neural networks? In: Proceedings of the 7th International Conference on Learning Representations. New Orleans, USA: OpenReview.net, 2019. [28] Veličković P, Cucurull G, Casanova A, Romero A, Lio P, Bengio Y. Graph attention networks. In: Proceedings of the 6th International Conference on Learning Representations. Montréal, Canada: OpenReview.net, 2018. [29] Li J, Rong Y, Cheng H, Meng H L, Huang W B, Huang J Z. Semi-supervised graph classification: A hierarchical graph perspective. In: Proceedings of the World Wide Web Conference. San Francisco, USA: ACM, 2019. 972−982 [30] Duvenaud D, Maclaurin D, Iparraguirre J, Bombarell R, Hirzel T, Aspuru-Guzik A, et al. Convolutional networks on graphs for learning molecular fingerprints. In: Proceedings of the 28th Annual Conference on Neural Information Processing Systems. Montreal, Canada: MIT, 2015. -

 下载:
下载:



计量
- 文章访问数: 1569
- HTML全文浏览量: 1084
- PDF下载量: 255
- 被引次数: 0
