

Identifying Candidate Disease Genes in Multilayer Heterogeneous Biological Networks
-
摘要: 现有大多数用于识别候选疾病基因的随机游走方法通常优先访问高度连接的基因, 而可能与已知疾病有关的不知名或连接性差的基因易被忽略或难以识别. 此外, 这些方法仅访问单个基因网络或各种基因数据的聚合网络, 导致偏差和不完整性. 因此, 设计一种能控制随机游走运动方向和整合多种数据源的候选疾病基因识别方法将是一个迫切需要解决的问题. 为此, 首先构建多层网络和多层异构基因网络. 然后, 提出一种游走于多层网络和多层异构网络的拓扑偏置重启随机游走(Biased random walk with restart, BRWR)算法来识别疾病基因. 实验结果表明, 游走于不同类型网络上的识别候选疾病基因的BRWR算法优于现有的算法. 最后, 应用于多层异构网络上的BRWR算法能预测未诊断的新生儿类早衰综合征中涉及的疾病基因.Abstract: Most of existing random walk methods to identify candidate disease genes preferentially visit highly-connected genes, while unwell-known or poorly-connected genes probably relevant to known diseases are more easily ignored or complicated to identify. Moreover, these methods access only a single gene network or an aggregated network of various gene data, leading to bias and incompleteness. Therefore, it is a pressing challenge for controlling the motion direction of random walk and for integrating multiple data sources involving different information for disease-gene identification. To this end, we first construct a multilayer network and multilayer heterogeneous genetic network. Then, we propose a topologically biased random walk with restart (BRWR) algorithm applicable to multilayer and multilayer heterogeneous networks for the identification of candidate disease genes. Experimental results show that the BRWR algorithm to identify candidate disease genes outperforms the state-of-the-art ones on different types of networks. Finally, the BRWR algorithm on multilayer heterogeneous networks is used to predict disease genes implicated in the undiagnosed neonatal progeroid syndrome.
-
现代复杂机电系统的维修工作与传统装备的维修工作存在着较大的差异.传统的机电系统因其电气化程度低, 其结构和功能比较简单, 相对而言易于维修; 现代大型机电系统组成结构相对复杂、功能强大, 维修工作相对困难.现代复杂系统内部充满了各种信息数据, 像设备的参数信息、活动信息、状态信息, 系统管控的业务信息等, 这些流数据和离散数据通过利用电子信息技术得到了采集和保存.分析发现, 这些数据具备大数据的5Vs的特征:数据量大(Volume), 如10 000个测点、20维数据特征、每0.5秒采集一次、分辨率为32位的5年数据量约2×1 015b, 达到了P量级, 符合数据量大的特征; 速度快(Velocity), 如此多的采集点位和采集分辨率, 采集速度必须非常的快; 类型多(Variety), 除基本的数据类型外, 还有流数据、媒体、图像等类型; 有价值(Value), 利用大数据技术能从这些复杂的数据中找到有用信息, 为维护决策提供支持; 真实性(Veracity), 数据采集于现场等.于是, 大数据结构化和数据驱动的复杂系统维修决策方法的研究被提出.研究的大数据结构化利用了层次分析法(Analytic hierarchy process, AHP)对复杂系统进行分析, 将与决策相关的元素分解成目标、准则、方案等多个层次, 在此基础之上实现定性和定量的计算方法. AHP方法是20世纪70年代初美国运筹学家匹茨堡大学教授萨蒂提出的一种决策论, 它是在网络系统理论和多目标综合评价的基础上进行一种层次权重决策分析方法.现在, 随着物联网和大数据技术的发展, 各类海量的数据为人类活动提供了依据, 于是, 产生了数据驱动技术, 它为企业生产、经营、管理提供决策和依据.
传统技术的优点:针对性强, 故障特征明显, 使用范围比较具体; 主要采用的是信号处理技术; 数据来源于同一对象, 且为连续的同一类型数据; 此诊断技术涉及的学科相对比较专一.技术的缺点:只能对已发送故障进行诊断, 不能预测将要发现的故障, 对引起故障的外部因素不能做诊断, 针对性强, 对于认知不足的现象, 无法诊断. AHP方法的优点:应用范围广泛, 从单一设备到系统层级, 注重环境因素对设备造成的影响; 方法技术较为全面, 不仅能进行信号处理, 还可以进行逻辑推理、最优规划等智能技术; 数据来源广泛, 能够和大数据技术进行紧密结合; 方法涉及多专业、多学科的知识.方法的缺点:需要强大的知识支撑, 诊断存在精确度的问题, 和大数据技术类似, 方法尚处在一个发展完善的过程中.
近些年, 利用AHP进行大数据结构化的研究相对较少, 从吴信东等学者的“从大数据到大知识: HACE + BigKE”一文的综述中可见一斑[1], 但AHP方法在装备制造工程的应用却比较常见.如东北大学的刘强等提出了“过程工业大数据建模研究展望”, 对大数据给出了认知的概念和提出了要解决的问题[2]; 清华大学的卢兆麟等[3]结合自然语言处理和层次分析法有效和准确地评价了乘用车驾驶舒适性; 吉林大学田广东等[4]基于模糊AHP和灰色关联POPSIS实现了对产品拆解方案的评估研究; 南昌航天大学的秦国华等[5]通过AHP方法解决了夹具定位元件的选择问题; 东北大学的李强等[6]利用模糊综合层次评判法实现了精密齿轮制造工艺优化优先度的评判和排序.国外的伊朗德黑兰大学的Azadeh等[7]基于一致性层次分解法提升维修管理恢复性工程的评估水平; 波兰国家研究院的Podgórski利用AHP方法进行主导性能指标选择示例解决职业安全健康系统的运行性能评测问题[8].这些研究的一个共同特点就是使用AHP刻画复杂系统结构, 求解系统存在的一些难题, 本研究引入AHP方法实现复杂系统的大数据结构化问题.
数据驱动技术近几年出现了大量的研究成果.华南理工大学的姚锡凡等通过构建主动制造体现构架实现了一种大数据驱动的新型制造模式-主动制造[9]; 浙江大学和杭州电子科技大学的文成林等提出了基于数据驱动的微小故障诊断方法综述, 根据微小故障的特点探究微小故障诊断思想[10]; 上海交通大学和华中科技大学的张洁等针对智能车间制造数据呈现的大数据特性, 研究了大数据驱动的车间运行分析与决策方法体系[11]; 西安交通大学的雷亚国等通过深度学习利用机械频域信号训练深度神经网络, 摆脱对大量信号处理技术与诊断经验的依赖, 完成故障特征的自适应提取与健康状况的智能诊断[12]; 电子科技大学的彭卫文等通过性能退化试验设计和数据分析, 导出功能铣头的伪寿命数据, 应用贝叶斯方法构建融合试验信息和现场故障数据实现功能铣头可靠性的评估[13]; 西安交通大学和第二炮兵工程学院的张家良等[14]提出了基于非线性频谱数据驱动的动态系统故障诊断方法, 通过对一种非线性频谱特征提取, 利用最小二乘支持向量机分类器进行故障识别实现故障诊断; 北京交通大学的侯忠生等进行了数据驱动控制理论及方法的回顾和展望的研究, 详述了数据驱动控制理论和方法的适用条件, 并对对数据驱动控制理论的发展进行了展望[15].在国外, 如意大利米兰大学学者Grasso教授提出了一种增强振动信号分解的滚动轴承故障分析的数据驱动方法[16], 通过自动分解方法产生一个相关模式最小数, 形成一个共享类属性的固有模态, 这种增强模式没有任何信息损失, 把它作为滚动轴承故障分析的数据驱动方法; 比利时根特大学Janssens等通过多次使用随机梯度增强回归树方法实现基于数据驱动的海上风力发电机组多变量功率曲线建模[17].上述研究大多基于现实系统的数据进行系列研究方法, 即数据驱动方法.
目前, 关于大型设备或系统维修决策也是一个研究热点.如清华大学的周东华等发表了“工业过程异常检测、寿命预测与维修决策的研究进展”的论文, 为保障工业过程安全性、可靠性和经济性, 指出了该领域中存在的问题及未来的研究方向[18]; 浙江大学和德国诺丁汉大学的Zhang等提出基于周期性监测的牵引电机绝缘最优视情维修决策, 研究充分利用寿命信息, 并考虑冲击效应对维修策略的影响, 提出符合牵引电机绝缘运行特征的最优视情维修策略[19]; 工信部电子五所和华中科技大学的王远航等提出基于多故障模式的复杂机械设备预防性维修决策[20], 根据系统结构和功能特征进行关键故障模式辨识, 利用故障时间分布即寿命分布构建预防性维修决策的目标成本函数, 并通过遗传算法实现含整型约束的非线性优化问题求解; 太原科技大学的石慧等提出了考虑非完美维修的实时剩余寿命预测及维修决策模型[21], 建立了系统预防性维护的阈值变量和最小化平均维护费用的优化模型, 并采用微粒群算法求解.法国洛林大学的学者Medina-Oliva的团队分别提出了工业系统知识形式化的维修方法评估辅助决策[22]、视情维修决策的能效研究[23]、制造平台能效预测辅助维修决策研究[24]; 英国斯克莱德大学Iraklis等进行了增强检测、维护和决策的高级船运输系统状态监测研究[25].
从以上成果可以看到AHP和数据驱动技术在大型机电装备系统维修决策的研究较少, 并且关于复杂机电系统的维修决策研究是当前的一个热点.本文提出的大数据结构化与数据驱动的复杂系统维修决策方法旨在实现AHP、数据驱动和设备维修的结合.
1. 大数据的结构化
1.1 AHP建模
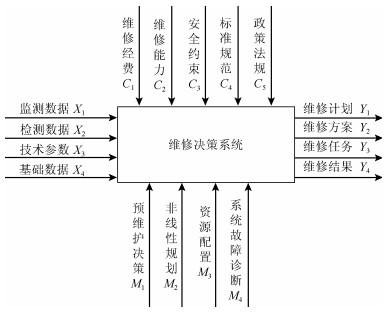
按照AHP的思想, 复杂系统可以理解为由输入(Input)、输出(Output)、控制(Control)及机制(Mechanism)等四类信息作用的系统, 据此建立了维修决策系统模型A, 如图 1所示.
1.2 数据定义
1) 输入(Input)数据
监测数据($ X_1 $), 指通过DCS对系统核心设备、重要指标或关键环节实时采集的数据;
检测数据($ X_2 $), 它是根据维修活动的管理和诊断的需要通过各种手段获取的有效数据.如日常巡检、点检、以及临时测试的数据;
技术参数($ X_3 $), 指技术改造、或者是技术变通措施的设计、重组、变更的数据;
基础数据($ X_4 $), 指系统设备资料、运行参数、设备耦合参数, 人员资料, 技术资料, 标准指标等数据.
分析看出, 数据$ X_1 $, $ X_2 $, $ \cdots $, $ X_n $代表了设备的各种状态量, 这些状态量都具有随机性, 即$ X_1 $, $ X_2 $, $ \cdots $, $ X_n $都是其状态的随机变量, 它们是随机分布的.
这里$ X $代表了设备单元某种状态在某个量值范围上下振荡的随机值.
对于整个维修系统, 可以看作为一个具有$ n $维向量维修输入的系统.
若定义系统的维修输入为$ X $, 那么, $ X_1 $, $ X_2 $, $ \cdots $, $ X_n $分别代表着$ n $个维修输入分量, 具有$ n $维分量时间$ T $的$ X $可写为$ X = (X_1, X_2, \cdots, X_n)^{\rm T} $.
定义, $ n $维向量$ X = (X_1, X_2, \cdots, X_m)^{\rm T} $的分布函数为$ F(x_1, x_2, \cdots, x_m) $, 记$ T = (t_1, t_2, \cdots, t_n)^{\rm T} $, $ b = (b_1, b_2, \cdots, b_m)^{\rm T} $, 则$ m \times n $维向量线性矩阵函数可记为式(1):
$$ \begin{equation} F(X) = (X_1, X_2, \cdots, X_m)^{\rm T} = kX+b \end{equation} $$ (1) 2) 控制(Control)数据
此部分指的是维修活动所受的人员、技术、费用、安全、规则等方面的限制和约束.
一是维修费用($ C_1 $), 维修活动所受到的资金限制.维修活动必需通过计划展开, 尽量避免临时性工作带来更多附加配备、人员劳务等方面的开销;
二是维修能力($ C_2 $), 指的是在现有的技术、设备、技术人员等状态下能否完成指定的维修任务;
三是安全约束($ C_3 $), 指保证人员、设备等方面不会发生的损失危害的情况下实施的系列活动;
四是标准规范($ C_4 $), 指活动必须满足一定条件的行业标准、规程规范;
五是政策法规($ C_5 $), 指维修活动一定要遵守国家的法令法规等内容.
与维修输入类似, 维修控制代表了对维修系统的另一类的输入, 这些输入量同样具有随机性, 并且是满足一定条件的分布函数.
定义1. 控制输入变量为$ C $, 那么, $ C_1, C_2, \cdots, C_n $分别代表着不同的输入分量, 具有$ n $维分量时间$ T $的$ C $记为$ C = (C_1, C_2, \cdots, C_n)^{\rm T} $.
维修控制输入$ C $实际是对系统维修的系列约束或限制性输入, 它与时间密切相关, 系统生命周期的不同时段, 其变量值是不同的, 对此可以建立$ n $维向量$ C = (C_1, C_2, \cdots, C_n)^{\rm T} $的分布函数为$ \Phi(c_1, c_2, \cdots, c_n) $, 记为, $ T = (t_1, t_2, \cdots, t_n)^{\rm T} $.
一般认为维修控制变量是满足0-1分布或二项分布的一系列随机值, 因此, 可做如下定义.
定义2.随机变量$ C $的分布函数为$ \Phi(c_1, c_2, $ $ \cdots, c_n) $, 且如满足0-1分布, 那么其分布函数可表示为式(2):
$$ \begin{equation} \Phi(C) = \Phi\{C = k\} = (p^k q^{1-k})^ {\rm T} \end{equation} $$ (2) 其中, $ k = 0, 1 $.
定义3.随机变量$ C $的分布函数为$ \Phi(c_1 $, $ c_2, \cdots, c_n) $, 且如满足二项分布, 那么其分布函数可表示为式(3):
$$ \begin{align} \Phi(C) = &\Phi\{C = k\} = (p^k q^{1-k})^{\rm T} = \\&(C(_n^k)p^k q^{n-k})^ {\rm T} \end{align} $$ (3) 其中, $ k = 0, 1, \cdots, n $.
3) 维修机制(Mechanism)
维修机制实质是基于系统的多种输入数据而进行各种数据运算与操作, 这些操作构成了系统处理的算法机制.
输入数据部分有结构化数据和非结构化数据, 在构造矩阵时, 需要进行混合数据处理, 在此维修机制部分定义了多种维修算子, 实现这些异构化数据间的运算和处理.
定义4. 假设系统维修机制变量为$ M $, $ M_1, M_2, \cdots, M_n $分别代表维修机制变量分量, 具有$ n $维分量时间$ T $的$ M $记为$ M = (M_1, M_2, \cdots, M_n)^{\rm T} $.那么维修机制函数$ G(M) $就可表示为式(4)的形式.
$$ \begin{equation} G = G(M) = G(A, B) = (A)*(B) \end{equation} $$ (4) 其中, $ A, B\in F(X) $或$ A, B\in\Phi(C) $, *为$ G(M) $的运算算子, 它是基于$ F(X) $, $ \Phi(C) $关系上的一种运算.按照维修功能的不同选择相应的维修机制$ G(M) $.此处定义算子为$ F(X) $和$ \Phi(C) $的广义积运算.
为了使矩阵的表示符合人们的习惯, 设$ F(X) $形成的矩阵为$ A = (a_{ik})_{m\times p}, \Phi(C) $形成的矩阵为$ B = (b_{kj})_{p\times n} $, 运算结果矩阵为$ C = (c_{ij})_{m\times n} $, 那么, 定义的广义积运算关系*为求和、求交、求积三种形式:
和计算如下式(5).
$$ \begin{equation} c_{ij} = \sum\limits_{k = 1}^p a_{ik}*b_{kj} \end{equation} $$ (5) 其中, $ i = 1, 2, \cdots, m; j = 1, 2, \cdots, n $.
交计算如下式(6).
$$ \begin{equation} c_{ij} = \bigcap\limits_{k = 1}^p a_{ik}*b_{kj} \end{equation} $$ (6) 其中, $ i = 1, 2, \cdots, m; j = 1, 2, \cdots, n $.
积计算如下式(7).
$$ \begin{equation} c_{ij} = \prod\limits_{k = 1}^pa_{ik}*b_{kj} \end{equation} $$ (7) 其中, $ i = 1, 2, \cdots, m; j = 1, 2, \cdots, n $.
预维修决策($ M_1 $), 应用概率估计、贝叶斯理论、灰色理论等对系统设备发生故障的可能性进行预测, 调节确定算子参数.
按照式(4), 可以得到下式(8).
$$ \begin{equation} G_1 = G(M_1) = G(A_1, B_1) = A_1*B_1 \end{equation} $$ (8) 非线性规划($ M_2 $), 系统中某些问题的出现规律不满足一般的线性模型, 需要用非线性的方式去建模处理, 遇到这一类现象时就调用非线性规划机制, 构建算子参数.
同理, 可以得到$ G_2 $的算子.
维修资源配置($ M_3 $), 运用一定的数学方法, 找出系统结构安全方面的薄弱环节, 对系统资源重组或是再分配, 使系统风险最小、可靠性最高运算方法.
类推, 可以得到$ G_3 $的算子.
系统故障诊断($ G_4 $), 利用分布式复杂机电系统故障传播、扩散机理, 分布式复杂机电系统风险源辨识及事故原因推理方法, 分布式复杂机电系统风险评估方法等研究对系统故障进行诊断, 并输出评判结果.
类推, 可以得到$ G_4 $的算子.
4) 输出(Output)数据
该部分概括为:维修计划($ Y_1 $), 维修任务($ Y_2 $), 维修方案($ Y_3 $), 维修实施结果($ Y_4 $)等内容.
定义输出变量为$ Y $, 它是维修系统和维修机制作用的结果.那么, 具有$ n $维分量时间$ T $的维修输出$ Y $可写为$ Y = (Y_1, Y_2, Y_3, \cdots, Y_n)^ {\rm T} $.
如果看作输出为一组随机变量的集合$ Y(Y_1, Y_2, Y_3, \cdots, Y_n) $, 则$ Y $是变量$ F $, $ \Phi $通过$ G $作用的结果, 那么有下式(9):
$$ \begin{align} Y = &Y(F, \Phi, G) = \\& \{F(X); \Phi(C); G(M)|G(F(X), \Phi(C))\} = \\& G(F(X)* \Phi(C)) \end{align} $$ (9) 2. 数据驱动的模型设计
2.1 AHP的数据驱动
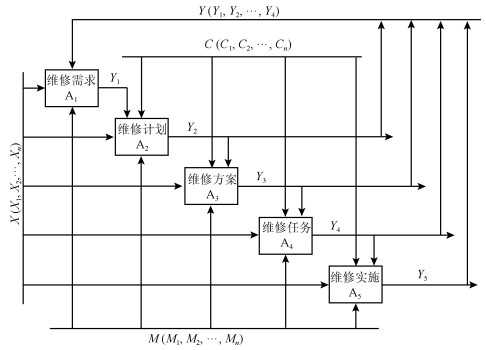
设计思想是利用AHP方法对目标系统进行分解, 产生下一层级相互独立、相互平行的功能模块, 考察每个模块和系统输入数据的相关性, 研究各模块在大数据作用下的作用机理, 建立数据变量变化的作用函数关系.维修决策系统模型A进行分解可得到下一级维修模型A-1, 如图 2所示.维修决策系统可以划分为5个功能模块:维修需求(A$ _1 $)、维修计划(A$ _2 $)、维修方案(A$ _3 $)、维修任务(A$ _4 $)、维修实施(A$ _5 $).
根据现代控制理论方法, 通过式(1)、(2)形成的结构化数据, 建立起来源于大数据变量的数据矩阵, 结合数据矩阵的计算方法, 设计出数据驱动的维修决策模型.基于式(9), 就可以得到维修决策数据驱动模型函数式(10).
$$ \begin{align} Y_i = &Y(F_i, \Phi _i, G_i) = \qquad\qquad\quad \\& \{F(X_i); \Phi(C_i); G(M_i)|G_i(F(X_i), \Phi(C_i))\} = \\& G_i(F(X_i)* \Phi(C_i)) \end{align} $$ (10) 维修需求(A$ _1 $), 依据系统工作的状态数据$ X_1, X_2, X_3, X_4 $, 借助于系统维修机制, 结合维修控制的约束, 预测系统设备故障的发生以及发生的时间, 给出关于系统健康状况的一个评测报告, 产生一系列设备维修需求的结果集.
依照式(10), 可以得到维修需求的数据驱动算子, 如下式(11).
$$ \begin{equation} Y_1 = Y(F_1, \Phi _1, G_1) = G_1(F(X_1)* \Phi(C_1)) \end{equation} $$ (11) 维修计划(A$ _2 $), 在维修需求的基础上, 综合维修输入、维修控制等因素, 经过信息的分析、加工与处理, 然后确定维修对象、维修内容、维修级别、维修时段, 判断所需维修人员支持、物质材料的供应、所需设备工具的有无、所需资金的配备、维修技术的储备、以及环境法律法规的约束等方面的可行性, 最终形成维修计划输出的过程.同上, 得到$ Y_2 $的计算算子.
维修方案(A$ _3 $), 根据维修计划详细设计维修活动的过程, 核心是维修技术的配置.通过对维修输入、维修机制、维修控制等方面的综合考虑, 实际中主要考虑维修时间、维修方法、以及维修资金这三种因素, 从而产生系列维修方案, 然后, 从中选择一个可行的最佳方案.同上, 可以得到$ Y_3 $的计算算子.
维修任务(A$ _4 $), 它是维修方案与维修人员的结合.在维修控制的限制条件下, 对维修活动所进行的人力资源配置和调度.从另一个角度讲, 确定某些人员实施某项维修方案就是下达维修任务.一个公司通常是由多个从事不同工种的人员组成, 大家按照不同的分工协同配合才能顺利地完成维修任务.同上, 可以得到$ Y_4 $的计算算子.
维修实施(A$ _5 $), 基于上述的维修任务, 维修人员按照不同的时序进行设备维修的过程.此部分非常注重维修过程控制, 像维修技术的培训、物质材料的领取、设备工具的配备、作业实施等.另外, 这项工作的质量管理相当重要, 像执行过程的记录、验收方法、验收过程、实施结果记录、质量验收记录等都必须严格控制.同上, 得到$ Y_5 $的计算算子.
2.2 AHP与数据驱动的迭代
此部分的设计思想是把AHP方法进行迭代应用, 把任一子模块分解为相互独立、相互平行的多个下一级子模块, 对大数据与下一级子模块进行分析, 建立数据变量及其相应的作用函数.
通过AHP和数据驱动方法的迭代应用, 维修模型A-1的维修方案A$ _3 $子模块被分解为检修技术(A$ _{31} $), 安全措施(A$ _{32} $), 备品备件(A$ _{33} $), 方案管理(A$ _{34} $)四个下一级子模块, 如图 3所示.
那么, 按照现代控制系统理论, 维修方案A$ _3 $子系统(模块)的数据驱动维修决策可以使用下式(12)表示.
$$ \begin{align} y_{3i} = &y(f_{3i}, \phi _{3i}, g_{3i}) = \\& \{f(x_{3i}); \phi(c_{3i}); g(m_{3i})|g_{3i}(f(x_{3i}), \phi(c_{3i}))\} = \\& g_{3i}(f(x_{3i}), \phi(c_{3i})) \end{align} $$ (12) 检修技术(A$ _{31} $), 提取检修计划项, 对各项检修内容、技术要求做明确的审定.由于设备异常的多样性, 必需严格按照技术要求详细地列出检修操作的每一步, 以及是否达到指标要求的确认.
基于数据驱动的决策分量$ y_{31} $可用式(13)表达.
$$ \begin{align} y_{31} = &y(f_{31}, \phi _{31}, g_{31}) = \\& \{f(x_{31}); \phi(c_{31}); g(m_{31})|g_{31}(f(x_{31}), \phi(c_{31}))\} = \\& g_{31}(f(x_{31})* \phi(c_{31})) \end{align} $$ (13) 安全措施(A$ _{32} $), 依据检修内容和技术要求, 涉及的操作安全项必须一一列举出来, 每项必须有具体安全操作步骤、注意事项, 强调有严格的操作顺序、顺序确认等, 如动火类的安全操作需要进行动火分析, 记录分析结果和分析人员确认等要求.安全事项完成后, 再做一次安全性检查, 填写检查结果, 记录检查人员并签字.同样, 可以得到决策分量$ y_{32} $的表达.
备品备件(A$ _{33} $), 维修计划和维修实施中需要备品备件, 维修系统中要有备品备件的申购功能, 备品备件的领用记录功能.按照预维修展开的维修活动, 备品备件必需通过科学的预测和物流机制活动进行管理, 最为理想做法是满足生产正常运行需求而零库存.备品备件的管理直接影响企业的生产成本, 必需做好备品备件的信息共享和流通记录工作, 为企业管理提供最直接的数据支持.同样, 可以得到决策分量$ y_{33} $的表达.
方案管理(A$ _{34} $), 是维修方案的重要环节, 是对维修方案最后输出的确认, 它是对维修活动的一种约束, 保障了维修活动的正确有序进行.方案审核要求的操作有确认、修改、取消等.同样, 可以得到决策分量$ y_{34} $的表达.
维修系统经过A-2模型的功能分解, 系统模型基本上能够满足维修功能的工程化要求了.把模型A-1中的每个模块都可做类似的分解建模, 整个机电系统的维修功能建模算是完成了.建模中必须注意, 机电系统维修功能活动的五个部分相互联系、相互制约, 系统须保持完整性、一致性和严密的逻辑性.这种基于AHP的机电系统功能建模形成的设备维修体系是科学、严密和实用的, 非常符合大型生产的维修实际情况.
从图 3可以看出, A$ _{33} $部分的数据驱动计算仅与变量$ C_1, C_4, M_3 $和$ Y_2 $有关, 在进行驱动运算时, 可以定义其他不相关的变量为$ NULL $, 并定义$ NULL $计算的结果也为空.
3. 例子说明
3.1 系统描述
某一大型生产系统, 其设备连接如图 4所示, 图中标注了一些关键设备的运行状态监测点位, 点位测量数据值与设备的对应关系.现以此为例介绍大数据结构化与数据驱动的复杂系统维修决策方法的应用过程.
问题:某一次系统发生意外停机.
分析:此问题属于图 3中“检修分析与技术要求”的功能应用.按照文章思想, 先构建运行期间$ T $的数据结构化矩阵, 再利用维修机制算子$ Y_{31} $的输出式(13)进行计算, 最后对结果进行分析, 并输出结果$ Y_{31} $.
3.2 大数据结构化的构建
依照AHP的数据结构化方法对监测数据进行处理, 由于数据量非常庞大, 研究可以采用数据“帧”的方式进行处理, 这里的帧代表了当前被处理的数据数量, 也称之为数据窗口, 通过对当前数据的处理, 实现对帧数据的判断和评价.由于数据有很多连续的帧, 处理完当前帧, 然后转向下一帧.每次评判的结果, 作为维修决策使用的依据.
表 1是对某机组采集的一段截尾数据, 表中展示了10个采集点的12个数据值.
表 1 某设备群现场数据Table 1 A field data of equipment groups油压(MPa) 温度(℃) 气压(Kpa) 气流(kNm3/h) 液位(%) 汽压(Mpa) 箱振动(µm) 气温(℃) 气压(Mpa) 转速(rpm) 0.2125763 29.54823 95.59524 129.4939 46.18437 10.03053 20.73275 13.55311 0.5045177 11 182.2 0.2124542 29.60927 95.59524 129.2410 46.00122 10.03663 19.95911 13.73626 0.5045177 11 182.2 0.2128205 29.54823 95.59524 129.4476 46.18437 10.03053 19.95911 13.55311 0.5045177 11 182.2 0.2126984 29.54823 95.59524 129.5999 46.21490 10.03663 20.15137 13.55311 0.5045177 11 182.2 0.2126984 29.54823 95.59524 129.6462 46.15385 10.03663 20.10559 13.55311 0.5045177 11 182.2 0.2124542 29.60927 95.59524 129.1827 46.18437 10.03053 20.39399 13.73626 0.5045177 11 182.9 0.2123321 29.54823 95.59524 129.9566 46.21490 10.02442 20.00946 13.55311 0.5047619 11 182.9 0.2125763 29.54823 95.59524 130.0745 46.27595 10.02442 19.56999 13.55311 0.5045177 11 184.3 0.2122100 29.54823 95.55556 129.5866 46.27595 10.02442 19.71648 13.73626 0.5045177 11 184.3 0.2125763 29.54823 95.55556 129.7047 46.27595 10.02442 20.54048 13.55311 0.5045177 11 184.3 0.2126984 29.54823 95.59524 129.6329 46.33700 10.01832 19.90875 13.73626 0.5042735 11 183.6 0.21221 29.54823 95.55556 129.9104 46.39805 10.01221 19.90875 13.55311 0.5045177 11 185.0 按照大数据结构化的设计要求, 表 1中数据被AHP划分为决策系统的维修输入数据.应用系统维修输入数据结构化式(1)建立维修状态输入的结构化数据, 那么定义$ t $时刻设备的运行状态数据结构化矩阵$ A $, 即取当前帧连续的10行数据, 得到矩阵$ A $如下所示:
$$ A = \left[ { \begin{array}{ccccc} 100 800 & 0.2125763 & 46.18437 & 131.8363 & 11 155.87 \\ 100 810 & 0.2124542 & 46.00122 & 132.5042 & 11 156.56 \\ 100 820 & 0.2128205 & 46.18437 & 132.2923 & 11 155.87 \\ 100 830 & 0.2125763 & 46.18437 & 131.3540 & 11 163.48 \\ 100 840 & 0.2126984 & 46.21490 & 132.0667 & 11 161.41 \\ 100 850 & 0.2124542 & 46.27595 & 131.9528 & 7 232.401 \\ 100 900 & 0.2123321 & 46.39805 & 131.8680 & 1 966.924 \\ 100 910 & 0.2125763 & 46.27595 & 58.93486 & 625.5474 \\ 100 920 & 0.2122100 & 46.33700 & 0 & 75.64297 \\ 100 930 & 0 & 0 & 0 & 0 \\ \end{array}} \right] $$ 接下来, 对维修控制数据进行结构化处理, 依照式(2), 建立$ t $时刻系统控制输入矩阵$ B $, 结果如下所示:
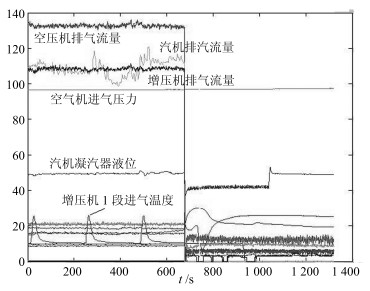
$$ B = \left[ \begin{array}{c} 100 850 \pm 50 \\ 0.3 \pm 0.2 \\ 46 \pm 2 \\ 132 \pm 2 \\ 11 160 \pm 5 \\ \end{array} \right] $$ 为了保障系统安全运行, 企业通常都采用了DCS系统对重要运行指标进行监控.根据监控数据, 维修功能就能够实时判断系统运行的平稳性, 并通过对系统设备状态的控制与调整保持设备运行的平稳性, 像系统故障产生时对故障源进行分析和查找.图 5显示了DCS采集压缩机组多个点位的状态变化情况, 它是系统设备运行状态的时序图.图中分别标注了不同设备的状态变化曲线, 并使用了不同的颜色对设备状态进行了区分.
3.3 数据驱动算子及应用
接下来利用维修功能模型并结合监控设备状态数据介绍系统设备异常时的维修方法, 即利用研究方法实现系统故障的诊断方法.
这里给出一组现场数据的运行状态实例.一次事故中伴随着一声异响全部机组都停止了运行, 监控信息显示空压机由于高位阀指令突然由76的开度自动增至98的开度, 蒸汽流量由188T降至12T, 空压机转速由11 149转降至3 700转, 此时, 操作人员采取了紧急停车的处理措施, 设备异常状态变化情况, 如图 5所示.
检修过程.停车后根据监测的转速、油压的变化趋势情况, 首先对FT7623流量变送器进行了检查测试, 结果确认为正常; 接着对高位阀V-15进行功能性测试, 当操作人员给出指令后, 发现高位阀不动作; 进一步对信号接线端子进行检测, 发现信号没有传送到电液转换器上, 初步判断信号电缆故障; 结合以往故障分析, 发现此前空压机透平轴密封圈泄漏, 高温气体辐射到信号电缆上, 导致仪表电缆保护套管被烤坏, 信号传输电缆也被烤坏, 最终发生信号故障, 导致高位阀无法动作, 机组工艺操作工手动停车.
接下来对系统进行分析.从监控设备数据的状态趋势、以及试验结果分析, 确认为当时的信号传输线出现短路, 控制信号丢失, 自动调节流量开关功能失效导致系统运行异常发生, 并最终引起空压机停车事故.拆线检查确认电缆由于被烤导致线路故障, 维修采取了将信号电缆用铁丝吊起, 远离泄漏部位, 下部采取用石棉板隔离的方法进行保护.
图 5显示了在发生故障时, 空压缩机pSE7655、pSE7656、pSE7657的转速由11 149转直接降至3 700转的状态曲线图, 以及空气压缩机排气流量(A_aFI7601)由188T降至12T的状态曲线图.
空压机A_aFI7601排气流量的监控数据如表 2, 它是事故发生的现场数据, 也是24小时的记录数据, DCS系统是每10秒钟进行一次数据采集.
表 2 测点设备的流量数据Table 2 The flux datum of measuring pointsf(Ti+1) f(Ti+2) f(Ti+3) f(Ti+4) f(Ti+5) f(Ti+6) f(Ti+7) f(Ti+8) f(Ti+9) f(Ti+10) 131.4101 131.5507 131.3375 131.6185 132.0129 133.0345 131.6311 132.4577 132.7883 132.7623 132.6695 131.5038 132.1837 131.4313 131.8313 132.5042 132.2923 131.354 132.0667 131.9528 131.868 58.93486 0 0 0 0 0 0 0 0 空压缩机pSE7655、pSE7656、pSE7657的转速监控数据值见表 3, 数据记录和采集方式给出了事故发生的$ T $周期数据, 即24小时的记录数据, DCS系统是每10秒钟进行一次数据采集.
表 3 测点设备的转速数据Table 3 The speed datum of measuring pointss(Ti+1) s(Ti+2) s(Ti+3) s(Ti+4) s(Ti+5) s(Ti+6) s(Ti+7) s(Ti+8) s(Ti+9) s(Ti+10) 11 159.33 11 157.25 11 156.56 11 157.95 11 162.79 11 159.33 11 156.56 11 155.87 11 156.56 11 155.87 11 163.48 11 161.41 7 232.401 1 966.924 625.5474 75.64297 0 0 0 0 0 33.64624 33.81691 33.98817 0 0 0 0 0 0 在上述模型基础上对$ T $时间段内系统状态进行维修评价.首先按照系统研究模型取数据帧作为基础数据, 然后对这些数据进行广义积运算, 得到一个新的矩阵, 该矩阵的值就代表了系统运行状态.
按照式(5)、(6)和(7)有下式(14).
$$ \begin{align} &Y_i(t) = \{F_i(X); \Phi _i(C); G_i(M)|(F_i(X), \Phi_i(C))\} = \\ &\quad \{A\ast B|(\min\Phi_i (C)\leq F_i (X)\leq\max\Phi_i (C))\vee \quad \\ &\quad (F_i (X)\wedge\Phi_i (C))\vee(F_i (X)\ni\Phi_i (C))\} \end{align} $$ (14) 运算算子*为矩阵的广义乘, 保持被乘矩阵行数与乘矩阵的列数相同, 对应的数值进行比较运算, 符合比较规则取值为1, 不符合规则取值为0, 最终项取行列运算值的交计算, 最后得到一个0-1矩阵, 计算过程如下式(15)、(16).
计算方法依照式(14)推出输出矩阵项的求解式(15)和式(16).
$$ \begin{equation} c_{ij}(t) = \left\{ \begin{array}{ll} 1, &\mbox{若}\; a_{ij}\leq b_{ji} \\ 0, & \mbox{否则} \\ \end{array} \right. \end{equation} $$ (15) 进一步, 依照式(15)可以推出输出计算矩阵式(16).
$$ \begin{equation} c_{ij} (t) = \left\{ \begin{array}{ll} 1, \mbox{若}\; (t_i\leq t)\wedge(f_1 (t_i )\leq \Phi_1 (t))\\ \qquad \cdots\wedge(f_n (t_i )\leq \Phi_n (t)) \\ 0, \mbox{否则} \\ \end{array} \right. \end{equation} $$ (16) 上述例子的求解结果即是:
$$ \begin{align} &c_{11} (10) = (b_{11}\in a_{11})\cap (b_{21}\in a_{12})\cap \quad \\ &\quad (b_{31} \in a_{13})\cap (b_{41}\in a_{14})\cap (b_{51}\in a_{15}) = \\ &\quad (1)\cap (1)\cap (1)\cap (1)\cap (1) = 1 \end{align} $$ 同理得到其他各项:
$$ \begin{align*} c_{21} (10) = 1 \nonumber \\ \vdots \qquad \nonumber \end{align*} $$ $$ \begin{align*} c_{61} (10) = 0 \nonumber \\ \vdots \qquad \nonumber \end{align*} $$ 按照上述算法得到故障值矩阵$ C $如下:
$$ c_{10} = \left[ 1\; \; 1\; \; 1\; \; 1 \; \; 1 \; \; 0\; \; 0 \; \; 0\; \; 0\; \; 0 \right]^{\rm T} $$ 在矩阵$ C $中, 1代表了检测数据正常, 0说明系统异常.对于异常项可以使用类似的矩阵运算算子定位系统异常单元.
按照矩阵$ C $值进行节点状态检查, 发现对应节点V-15状态异常时, 测试确认其状态失灵, 即不能按照指令要求对电磁阀的开度进行调节; 进一步检修发现控制信号不能到达电磁阀, 检查从外观上看电磁阀传输线有烤焦现象; 更换传输信号线, 结果故障排除.最终确认系统异常由电磁阀V-15失效所致, 并认为V-15节点是引起V-16节点异常的故障源.
对比于人工方法查找故障, 可以发现研究的系统维修功能模型具有快速、简洁、高效、实用的特点, 能够满足实际查找故障的要求, 精确找到故障源问题节点.可以为企业安全生产提供切实有效的系统维修帮助.另外, AHP的功能建模方法是基于统计数据的解决方法, 通过对数据分析求解问题, 数据来源越丰富, 求解问题也就越准确. AHP和数据驱动的设备维修决策方法可以作为分布式复杂机电系统事故预防的指导策略, 提高系统的可靠性与安全性.
4. 总结
4.1 传统技术与AHP方法的对比
1) 应用的范围不同
传统技术是以机电设备为对象进行的系列故障诊断和维护技术, 它通过对设备的回旋往复运动单元进行信号采集和处理, 再根据信号曲线特性推理和判断设备部件发生的故障情况, 最后实施相应的维修决策的过程.传统技术的故障诊断主要是针对单个设备进行的.
AHP方法是以机电系统为对象开展故障诊断和维护维修决策的, 机电系统由众多机电设备组成, 某个设备故障现象可能是由相邻设备所引起.解决这里问题常是对机电系统进行建模, 然后基于模型展开相应的故障推理诊断工作. AHP方法重在研究设备环境对其造成的危害, 结合大数据技术是非常合适的, 它是对传统技术的扩充和扩展.
2) 使用的方法不同
传统技术的故障诊断是对机电设备回旋运动装置进行特征信号采集和提取, 再根据其特征信号类型判断设备的故障类别和等级, 最后进行相应的维修维护.其关键是信号处理, 常采用傅里叶变换、小波变换等方法对信号进行处理.特征信号有八字型、香蕉型、月牙型、锯齿型、非同心圆等, 代表了偏心、不对中、磨损、油膜振荡等故障.如下图 6所示.诊断核心方法就是傅氏变换, 函数形式为式(17).
$$ \begin{equation} F(X) = \digamma[f(t)] = \int _{-\infty}^\infty f(t){\rm e}^{-{\rm i}\omega t}{\rm d}t \end{equation} $$ (17) AHP方法通过对机电系统的建模, 层层分解系统为不同功能单元模块, 同时找出影响因素、定义诊断机制、制定控制方法.诊断关键是如何抽象系统为相应的模型, 以及诊断推理方法、诊断机制.文中采用了数据结构化的构建方法, 定义了多种诊断机制模型, 形成结构化矩阵, 通过设计矩阵运算, 产生运算结果数据, 根据结果数据制定系统的维护决策.常见的系统优化路径如图 7所示.
代表性的系统优化方法有最优路径法, 其表达形式为式(18).
$$ \begin{equation} \begin{cases} \min\limits _{x\in {\bf R}^n}F(x) = \min\limits _{x\in {\bf R}^n} [f_1(x), f_2(x), f_3(x), f_4(x)]^ {\rm T}\\ {\rm s.t.} \quad g_j(x)\geq 0, \qquad j = 1, 2, \cdots, m \\ \qquad h_k(x)\geq 0, \qquad k = 1, 2, \cdots, p \end{cases} \end{equation} $$ (18) 3) 数据源不同
传统技术的数据来源机电设备的时序信号, 是基于时间的纵向信号, 可以是一维数据、二维数据或三维数据, 这些数据必须连续不间断. AHP方法的数据来源比较复杂, 类型也比较多, 就是所谓的大数据, 主要是利用信号间的联系和相关性, 辨识设备的异常变化, 此方法更注重于数据广度, 注重环境对设备的影响, 数据源相对越全面越好.
4) 涉及的学科不同
传统技术主要使用信号处理技术, 对信号进行变换, 提取设备工作中的特征信号. AHP方法不仅涉及到信息处理技术、概率论的使用, 另外, 离散数学、图理论也在AHP方法中广泛应用.此方法既能解决系统出现的线性问题, 也能解决系统中存在的非线性问题.
4.2 结论
通过上述四个方面的比较分析, 可知AHP方法优越于传统的故障诊断方法.
文章提出了大数据结构化与数据驱动的复杂系统维修决策方法, 方法具有较强的创新性和实用性, 为大型工业系统的维修工作提供一个较好的思路.通过AHP和数据驱动对系统建模, 使维修活动变得结构合理、层次清晰、维修简洁, 理清了各种复杂的维修活动关系, 提高了维修效率.维修决策中, 定义了多个基于数据驱动的维修决策函数, 为维修活动提供了理论支持, 对企业的维修活动起到了很好的指导和帮助, 并能促进企业维修水平的提高.在使用基于AHP和数据驱动的设备维修决策方法中, 更加注重系统大数据的结构化和数据驱动思想的研究, 对于维修活动影响较大数据驱动方法的各种运算相对较少, 这将是未来的一个研究方向.
-

图 1 多层网络、异构网络、多层异构网络以及探索它们的随机游走路径(箭头的实线)的示意图
Fig. 1 Schematic of multilayer, heterogeneous and multilayer heterogeneous networks, together with paths of random walks (arrow solid lines)

图 2 非异构基因网络上不同方法的ROC曲线及其对应的AUC值
Fig. 2 ROC curves and AUC values of different algorithms on the non-heterogeneous gene networks

图 3 异构基因网络上不同方法的ROC曲线及其对应的AUC值
Fig. 3 ROC curves and AUC values of different algorithms on the heterogeneous gene networks

图 4 排名随偏置参数$ b $变化的累积分布
Fig. 4 The cumulative distributions of the ranking with change of the biased parameter $ b $

图 5 排名随参数变化的累积分布
Fig. 5 The cumulative distributions of the ranking with change of the parameters

图 7 所有偏置参数为 −5时的网络表示
Fig. 7 Network representation when all the biased parameters are −5

图 8 所有偏置参数为 −1时的网络表示
Fig. 8 Network representation when all the biased parameters are −1

图 10 所有偏置参数为1时的网络表示
Fig. 10 Network representation when all the biased parameters are 1
表 1 表型、基因和聚合网络的统计属性
Table 1 Statistical properties of phenotype, gene and aggregated networks
网络 节点数 边数 平均度 COEX 10 415 998 712 47.44 PPI 12 893 70 141 7.73 PATH 10 966 274 051 13.47 聚合网络 17 611 1 342 703 25.79 表型网络 7 324 29 853 4.38  下载: 导出CSV
下载: 导出CSV
表 2 不同的非异构网络上的不同方法的AUC值(%)
Table 2 AUC values of different algorithms on different non-heterogeneous networks (%)
PPI COEX PATH Aggregated Multilayer RWR 73.35 72.84 74.43 76.53 77.98 ProDige 79.12 73.63 80.29 83.27 84.12 NDOS 78.27 74.78 79.86 84.49 87.95 DRS 78.93 74.94 80.87 84.78 88.45 BRIDGE 79.91 74.26 81.51 85.13 89.33 BRWR 81.15 75.20 84.18 86.73 90.17
下载: 导出CSV
表 3 不同异构网络上的不同方法的AUC值(%)
Table 3 AUC values of different algorithms on different heterogeneous networks (%)
PPIH COEXH PATHH AggregatedH MultilayerH CIPHER 74.52 73.51 78.30 77.89 78.31 RWRH 80.37 75.34 79.47 83.67 86.53 MAXIF 80.91 76.56 80.15 84.02 88.43 LapRWRH 81.91 77.80 80.90 84.93 88.78 NRWRH 81.36 78.38 82.70 86.56 89.36 IDLP 82.08 79.25 83.37 87.79 90.16 BRWRH 82.36 80.91 85.17 89.65 91.09
下载: 导出CSV
-
[1] Guala D, Sonnhammer E L L. A large-scale benchmark of gene prioritization methods. Scientific Reports, 2017, 7: Article No. 46598 doi: 10.1038/srep46598 [2] Schwikowski B, Uetz P, Fields S. A network of protein-protein interactions in yeast. Nature Biotechnology, 2000, 18(12): 1257−1261 doi: 10.1038/82360 [3] Sharma V, Ranjan T, Kumar P, Pal A K, Jha V K, Sahni S, et al. Protein-protein interaction detection: Methods and analysis. Plant Biotechnology. New York: Apple Academic Press, 2018. 391−411 [4] Chen Y, Jiang T, Jiang R. Uncover disease genes by maximizing information flow in the phenome-interactome network. Bioinformatics, 2011, 27(13): i167−i176 doi: 10.1093/bioinformatics/btr213 [5] Zhang Y G, Wang Y, Liu J H, Liu X H, Hong Y X, Fan X, et al. IDLP: A novel label propagation framework for disease gene prioritization. In: Proceedings of the 22nd Pacific-Asia Conference on Knowledge Discovery and Data Mining (PAKDD). Melbourne, Australia: Springer, 2018. 261−272 [6] Chen Y, Wu X B, Jiang R. Integrating human omics data to prioritize candidate genes. BMC Medical Genomics, 2013, 6: Article No. 57 doi: 10.1186/1755-8794-6-57 [7] Lee J H, Zhao X M, Yoon I, Lee J Y, Kwon N H, Wang Y Y, et al. Integrative analysis of mutational and transcriptional profiles reveals driver mutations of metastatic breast cancers. Cell Discovery, 2016, 2: Article No. 16025 [8] Yang K, Zhao X Z, Waxman D, Zhao X M. Predicting drug-disease associations with heterogeneous network embedding. Chaos, 2019, 29(12): Article No. 123109 doi: 10.1063/1.5121900 [9] Yang A Y, Chen J Q, Zhao X M. nMAGMA: A network-enhanced method for inferring risk genes from GWAS summary statistics and its application to schizophrenia. Briefings in Bioinformatics, 2021, 22(4): Article No. bbaa298 doi: 10.1093/bib/bbaa298 [10] Köhler S, Bauer S, Horn D, Robinson P N. Walking the interactome for prioritization of candidate disease genes. The American Journal of Human Genetics, 2008, 82(4): 949−958 doi: 10.1016/j.ajhg.2008.02.013 [11] Li Y J, Patra J C. Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics, 2010, 26(9): 1219−1224 doi: 10.1093/bioinformatics/btq108 [12] Li Y J, Patra J C. Integration of multiple data sources to prioritize candidate genes using discounted rating system. BMC Bioinformatics, 2010, 11(S1): Article No. S20 doi: 10.1186/1471-2105-11-S1-S20 [13] Li Y J, Li J Y. Disease gene identification by random walk on multigraphs merging heterogeneous genomic and phenotype data. BMC Genomics, 2012, 13(S7): Article No. S27 [14] Xie M Q, Xu Y J, Zhang Y G, Hwang T, Kuang R. Network-based phenome-genome association prediction by bi-random walk. PLoS One, 2015, 10(5): Article No. e0125138 doi: 10.1371/journal.pone.0125138 [15] Zhao Z Q, Han G S, Yu Z G, Li J Y. Laplacian normalization and random walk on heterogeneous networks for disease-gene prioritization. Computational Biology and Chemistry, 2015, 57: 21−28 doi: 10.1016/j.compbiolchem.2015.02.008 [16] Valdeolivas A, Tichit L, Navarro C, Perrin S, Odelin G, Levy N, et al. Random walk with restart on multiplex and heterogeneous biological networks. Bioinformatics, 2019, 35(3): 497−505 doi: 10.1093/bioinformatics/bty637 [17] Doncheva N T, Kacprowski T, Albrecht M. Recent approaches to the prioritization of candidate disease genes. WIREs Systems Biology and Medicine, 2012, 4(5): 429−442 doi: 10.1002/wsbm.1177 [18] Yang K, Lu K Z, Wu Y, Yu J, Liu B Y, Zhao Y, et al. A network-based machine-learning framework to identify both functional modules and disease genes. Human Genetics, 2021, 140(6): 897−913 doi: 10.1007/s00439-020-02253-0 [19] Bonaventura M, Nicosia V, Latora V. Characteristic times of biased random walks on complex networks. Physical Review E, 2014, 89(1): Article No. 012803 [20] Ding C F, Li K. Centrality ranking in multiplex networks using topologically biased random walks. Neurocomputing, 2018, 312: 263−275 doi: 10.1016/j.neucom.2018.05.109 [21] Pio-Lopez L, Valdeolivas A, Tichit L, Remy É, Baudot A. MultiVERSE: A multiplex and multiplex-heterogeneous network embedding approach. Scientific Reports, 2021, 11(1): Article No. 8794 doi: 10.1038/s41598-021-87987-1 [22] Peng J, Zhou Y Y, Wang K. Multiplex gene and phenotype network to characterize shared genetic pathways of epilepsy and autism. Scientific Reports, 2021, 11(1): Article No. 952 doi: 10.1038/s41598-020-78654-y [23] Novoa-del-Toro E M, Mezura-Montes E, Vignes M, Térézol M, Magdinier F, Tichit L, et al. A multi-objective genetic algorithm to find active modules in multiplex biological networks. PLoS Computational Biology, 2021, 17(8): Article No. e1009263 doi: 10.1371/journal.pcbi.1009263 [24] Zhao B H, Hu S, Liu X E, Xiong H J, Han X, Zhang Z H, et al. A novel computational approach for identifying essential proteins from multiplex biological networks. Frontiers in Genetics, 2020, 11: Article No. 343 doi: 10.3389/fgene.2020.00343 [25] Dursun C, Smith J R, Hayman G T, Kwitek A E, Bozdag S. NECo: A node embedding algorithm for multiplex heterogeneous networks. In: Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Seoul, South Korea: IEEE, 2020. 146−149 [26] Bentley B, Branicky R, Barnes C L, Chew Y L, Yemini E, Bullmore E T, et al. The multilayer connectome of Caenorhabditis elegans. PLoS Computational Biology, 2016, 12(12): Article No. e1005283 doi: 10.1371/journal.pcbi.1005283 [27] Shi C, Li Y T, Zhang J W, Sun Y Z, Yu P S. A survey of heterogeneous information network analysis. IEEE Transactions on Knowledge and Data Engineering, 2017, 29(1): 17−37 doi: 10.1109/TKDE.2016.2598561 [28] Gómez-Gardeñes J, Latora V. Entropy rate of diffusion processes on complex networks. Physical Review E, 2008, 78(6): Article No. 065102 [29] Brin S, Page L. Reprint of: The anatomy of a large-scale hypertextual web search engine. Computer Networks, 2012, 56(18): 3825−3833 doi: 10.1016/j.comnet.2012.10.007 [30] Pan J Y, Yang H J, Faloutsos C, Duygulu P. Automatic multimedia cross-modal correlation discovery. In: Proceedings of the 10th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Seattle, USA: ACM, 2004. 653−658 [31] Smedley D, Köhler S, Czeschik J C, Amberger J, Bocchini C, Hamosh A, et al. Walking the interactome for candidate prioritization in exome sequencing studies of Mendelian diseases. Bioinformatics, 2014, 30(22): 3215−3222 doi: 10.1093/bioinformatics/btu508 [32] Kivelä M, Arenas A, Barthelemy M, Gleeson J P, Moreno Y, Porter M A. Multilayer networks. Journal of Complex Networks, 2014, 2(3): 203−271 doi: 10.1093/comnet/cnu016 [33] Rolland T, Taşan M, Charloteaux B, Pevzner S, Zhong Q, Sahni N, et al. A proteome-scale map of the human interactome network. Cell, 2014, 159(5): 1212−1226 doi: 10.1016/j.cell.2014.10.050 [34] Del-Toro N, Dumousseau M, Orchard S, Jimenez R C, Galeota E, Launay G, et al. A new reference implementation of the PSICQUIC web service. Nucleic Acids Research, 2013, 41(W1): W601−W606 doi: 10.1093/nar/gkt392 [35] Kanehisa M, Sato Y, Kawashima M. KEGG mapping tools for uncovering hidden features in biological data. Protein Science, 2022, 31(1): 47−53 doi: 10.1002/pro.4172 [36] Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Research, 2022, 50(D1): D687−D692 doi: 10.1093/nar/gkab1028 [37] Mi H Y, Muruganujan A, Casagrande J T, Thomas P D. Large-scale gene function analysis with the PANTHER classification system. Nature Protocols, 2013, 8(8): 1551−1566 doi: 10.1038/nprot.2013.092 [38] Schaefer C F, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, et al. PID: The pathway interaction database. Nucleic Acids Research, 2009, 37(S1): D674−D679 [39] Köhler S, Gargano M, Matentzoglu N, Carmody L C, Lewis-Smith D, Vasilevsky N A, et al. The human phenotype ontology in 2021. Nucleic Acids Research, 2021, 49(D1): D1207−D1217 doi: 10.1093/nar/gkaa1043 [40] Greene D, BioResource N I H R, Richardson S, Turro E. Phenotype similarity regression for identifying the genetic determinants of rare diseases. The American Journal of Human Genetics, 2016, 98(3): 490−499 doi: 10.1016/j.ajhg.2016.01.008 [41] Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, et al. Gene prioritization through genomic data fusion. Nature Biotechnology, 2006, 24(5): 537−544 doi: 10.1038/nbt1203 [42] Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, et al. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Research, 2017, 45(D1): D833−D839 doi: 10.1093/nar/gkw943 [43] Hanley J A, McNeil B J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology, 1982, 143(1): 29−36 doi: 10.1148/radiology.143.1.7063747 [44] Mordelet F, Vert J P. ProDiGe: Prioritization of disease genes with multitask machine learning from positive and unlabeled examples. BMC Bioinformatics, 2011, 12: Article No. 389 doi: 10.1186/1471-2105-12-389 [45] Wu X B, Jiang R, Zhang M Q, Li S. Network-based global inference of human disease genes. Molecular Systems Biology, 2008, 4: Article No. 189 doi: 10.1038/msb.2008.27 [46] Chen X, Liu M X, Yan G Y. Drug-target interaction prediction by random walk on the heterogeneous network. Molecular BioSystems, 2012, 8(7): 1970−1978 doi: 10.1039/c2mb00002d [47] Blatti C, Sinha S. Characterizing gene sets using discriminative random walks with restart on heterogeneous biological networks. Bioinformatics, 2016, 32(14): 2167−2175 doi: 10.1093/bioinformatics/btw151 [48] De Domenico M, Solé-Ribalta A, Gómez S, Arenas A. Navigability of interconnected networks under random failures. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(23): 8351−8356 [49] Pivnick E K, Angle B, Kaufman R A, Hall B D, Pitukcheewanont P, Hersh J H, et al. Neonatal progeroid (Wiedemann-Rautenstrauch) syndrome: Report of five new cases and review. American Journal of Medical Genetics, 2000, 90(2): 131−140 doi: 10.1002/(SICI)1096-8628(20000117)90:2<131::AID-AJMG9>3.0.CO;2-E [50] Kiraz A, Ozen S, Tubas F, Usta Y, Aldemir O, Alanay Y. Wiedemann-Rautenstrauch syndrome: Report of a variant case. American Journal of Medical Genetics Part A, 2012, 158A(6): 1434−1436 doi: 10.1002/ajmg.a.35336 [51] Kohl M, Wiese S, Warscheid B. Cytoscape: Software for visualization and analysis of biological networks. Data Mining in Proteomics: From Standards to Applications. Totowa: Humana, 2011. 291−303 [52] Becerra C H, Contreras-García G A, Perez Vera L A, Díaz-Martínez L A, Beltran Avendaño M A, Salazar Martínez H A. Wiedemann-Rautenstrauch syndrome prenatal diagnosis. Journal of Perinatology, 2014, 34(12): 954−956 doi: 10.1038/jp.2014.156 [53] Paolacci S, Bertola D, Franco J, Mohammed S, Tartaglia M, Wollnik B, et al. Wiedemann-Rautenstrauch syndrome: A phenotype analysis. American Journal of Medical Genetics Part A, 2017, 173(7): 1763−1772 doi: 10.1002/ajmg.a.38246 [54] Navarro C L, Esteves-Vieira V, Courrier S, Boyer A, Duong Nguyen T, Huong L T T, et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. European Journal of Human Genetics, 2014, 22(8): 1002−1011 doi: 10.1038/ejhg.2013.258 [55] Beauregard-Lacroix E, Salian S, Kim H, Ehresmann S, D'Amours G, Gauthier J, et al. A variant of neonatal progeroid syndrome, or Wiedemann-Rautenstrauch syndrome, is associated with a nonsense variant in POLR3GL. European Journal of Human Genetics, 2020, 28(4): 461−468 doi: 10.1038/s41431-019-0539-6 [56] Arboleda G, Ramírez N, Arboleda H. The neonatal progeroid syndrome (Wiedemann-Rautenstrauch): A model for the study of human aging? Experimental Gerontology, 2007, 42(10): 939−943 doi: 10.1016/j.exger.2007.07.004 [57] Zhao X M, Liu K Q, Zhu G H, He F, Duval B, Richer J M, et al. Identifying cancer-related microRNAs based on gene expression data. Bioinformatics, 2015, 31(8): 1226−1234 doi: 10.1093/bioinformatics/btu811 [58] He F, Zhu G H, Wang Y Y, Zhao X M, Huang D S. PCID: A novel approach for predicting disease comorbidity by integrating multi-scale data. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2017, 14(3): 678−686 doi: 10.1109/TCBB.2016.2550443 [59] Dong G Y, Feng J F, Sun F Z, Chen J Q, Zhao X M. A global overview of genetically interpretable multimorbidities among common diseases in the UK Biobank. Genome Medicine, 2021, 13(1): Article No. 110 doi: 10.1186/s13073-021-00927-6 期刊类型引用(12)
1. 周航旭,邵雯,李纬捷. 医院设备维修中的数据驱动决策与预测维护探讨. 中国设备工程. 2025(05): 61-63 .  百度学术
百度学术2. 刘雨蒙,郑旭,田玲,王宏安. 基于时序图推理的设备剩余使用寿命预测. 自动化学报. 2024(01): 76-88 . 本站查看3. 张欢. 基于大数据分析的石油仪表设备自动化维修决策支持系统研究. 中国石油和化工标准与质量. 2024(18): 43-45 . 百度学术4. 梁思远,周金浛,高占宝,于劲松,宋悦,张健. 机电系统健康状态预测和维修决策的双向优化方法. 仪器仪表学报. 2023(01): 131-142 . 百度学术5. 李天梅,司小胜,张建勋. 多源传感监测线性退化设备数模联动的剩余寿命预测方法. 航空学报. 2023(08): 94-112 . 百度学术6. 杨凯,张宇,原菊梅,张鑫,王银. 构建输电线路人工智能检测大系统. 系统科学学报. 2023(04): 120-125 . 百度学术7. 张友鹏,苏中集,石磊,张美艳. 基于嵌套粒子群结构的复杂系统维修决策优化方法. 计算机集成制造系统. 2023(11): 3800-3811 . 百度学术8. 刘沛津,王柳月,孙昱,史洁琳,晏东阳. 基于一致性哈希算法的分布式机电系统海量数据存储策略研究. 机床与液压. 2023(22): 31-38 . 百度学术9. 赵黎兴,侯兴明,徐兆文,贾超. 基于数据驱动的试验装备维修保障体系. 兵工自动化. 2022(07): 41-46 . 百度学术10. 李天梅,司小胜,刘翔,裴洪. 大数据下数模联动的随机退化设备剩余寿命预测技术. 自动化学报. 2022(09): 2119-2141 . 本站查看11. 张兵,苗琪琪,张株瑞,刘晓冰. 电机全生命周期质量大数据管控系统研究. 计算机应用与软件. 2022(11): 57-65 . 百度学术12. 李岳勋,吴挺文. 槽车外输智能管理业务系统建设. 电子技术与软件工程. 2020(12): 158-159 . 百度学术其他类型引用(15)
-
 下载:
下载:






计量
- 文章访问数: 822
- HTML全文浏览量: 429
- PDF下载量: 129
- 被引次数: 27
